In humans and other organisms, a single genome encodes development into distinct cell types and the responses of those cells to environmental conditions. Variation in how individuals respond to the environment is directly linked to survival and fitness. However, we still understand little about how these differences arise, are coded in the genome, and have been shaped by natural selection. My research seeks to answer these questions by combining comparative, population, and functional genomic data. In particular, I focus on changes in gene regulation – the mechanisms that control the timing, location, and amount that a gene is expressed – following stimuli that differ between individuals and species. Although my current projects focus on infectious disease, I am also interested in how the genome encodes appropriate responses to other environmental challenges such as social and energetic stress.
|
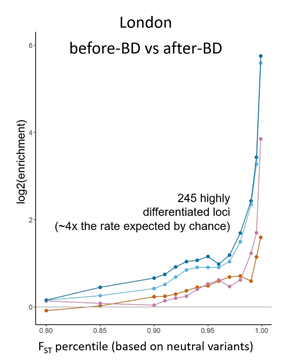
Natural selection on immune genes during the Black Death
Infectious diseases are among the strongest selective pressures in human evolution, but it is difficult to link specific diseases with genetic loci. My postdoc work has focused on adaptation during the single greatest mortality event in recorded history: the Black Death which was caused by Yersinia pestis. Using ancient genomic data, I identify an enrichment of positive selection among immune loci, including high-confidence variants which likely experienced positive selection. I then use in vitro assays and functional genomics (including single-cell RNA sequencing) to show several of these variants affect the expression of genes involved in antigen presentation and TLR signaling. Variation that was protective during the Black Death confers increased susceptibility to inflammatory and autoimmune disorders in modern individuals, representing the first empirical evidence connecting the selective force of past pandemics to present-day disease susceptibility. The first paper from this work has been published in Nature (Klunk & Vilgalys et al. 2022) and been profiled in Nature News & Views and by major news sources (including the NY Times, Science, CNN, and BBC ). We are continuing this line of research using single-cell genomics to identify genetic variation in the response to Y. pestis infection and whole-genome sequencing of ancient genomes.
|
|

|
Evolution of the immune response among nonhuman primates
Selective pressures on the immune system have generated broad differences in pathogen susceptibility, tolerance, and the immune response within and across species. For example, many diseases are less severe in their natural hosts, indicating that coevolution has produced mechanisms that limit the severity of infection. Changes in gene regulation are likely involved in these adaptations. However, our understanding of these changes, and the role they play as determinants of fitness, remains limited.
As a postdoc, I am starting two projects to investigate immune adaptations in primates. First, I am investigating adaptation to yellow fever. While many African primates are natural hosts and develop only mild symptoms, infection can be lethal in humans and other species. This project will use experimental in vitro challenges to compare the gene regulatory response to yellow fever infection between tolerant and susceptible primate species, and preliminary work is supported by a Pilot Research Award from the Emory National Primate Research Center. More generally, I am also beginning to examine within- and between-species variation in the immune response to bacterial and viral stimuli. In addition to detecting differences in the gene regulatory response between species I will map genetic variation that affects the response within species, providing complementary insight into the genes and regulatory pathways that affect disease susceptibility. This work is the focus for my K99 application which recently received a promising impact score. |
Dissertation Research
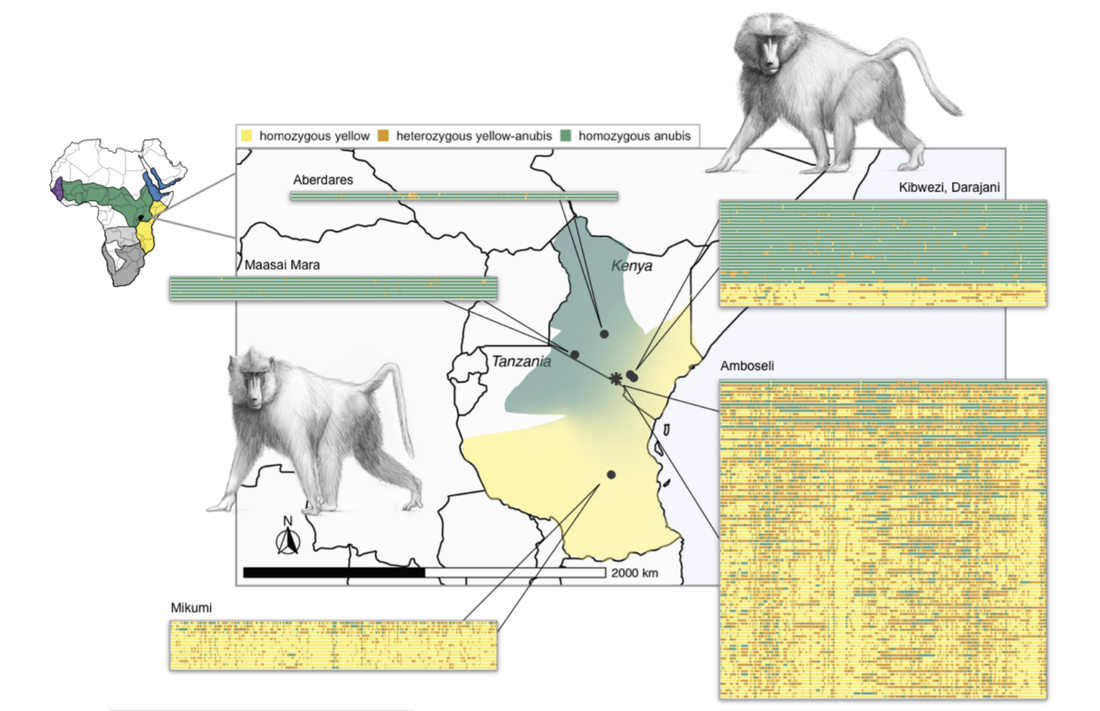
My dissertation research studied the evolution of gene regulation in baboons as a model for understanding the evolution of humans and other primates. Modern baboons (genus Papio) comprise 6 species which have spread across much of Africa over the past two million years. At the boundary between species, baboons often hybridize producing admixed offspring. The evolutionary history of baboons therefore mimics many aspects of human evolution, but with multiple extant species which can be studied in natural environments. Thus, baboons can serve as a natural model to understand phenomena in human evolution (such as admixture with Neanderthals and Denisovans) which we can no longer directly observe.
Selection on gene regulatory traits following admixtureAdmixture (the interbreeding of divergent, but related lineages) has been an important process in primate evolution. For example, admixture between modern humans and archaic hominins (such as Neanderthals and Denisovans) continues to affect genetic and phenotypic diversity among living people. However, the extinction of other hominin species thousands of years ago has limited insights into the immediate consequences of admixture in our own lineage.
Despite no major fitness costs to hybrids, we identify signatures of selection against introgression that are strikingly similar to those described for archaic hominins. These signatures are stronger where local ancestry affects gene expression, supporting the hypothesis that natural selection targeted large regulatory effects following archaic hominin admixture. This work has been published in Science (Vilgalys & Fogel et al. 2022). |
|
|
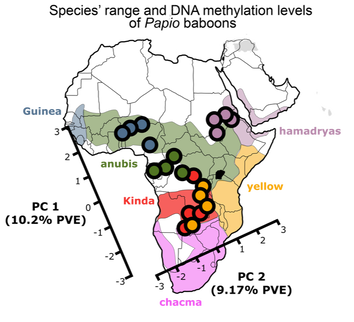
Evolution of DNA methylation among Papio baboonsDNA methylation is an epigenetic mark which chemically alters DNA without changing the underlying DNA sequence. DNA methylation serves as an important link between genetic variation and gene expression, which is also environmentally responsive and can remain stable across cell divisions. Evolutionary changes to DNA methylation likely contribute to the evolution of gene expression levels, and recent studies have provided a detailed picture of DNA methylation divergence between human populations and between humans and other great apes.
Here, I examine the relationship between genetic changes and divergence in DNA methylation using Papio baboons. Our results support the idea that divergence in DNA methylation is primarily explained by the nearby DNA sequence. Furthermore, unlike gene expression, DNA methylation appears to evolve most commonly via genetic drift rather than due to natural selection. The results from this project have been published in Molecular Biology and Evolution and can be found here. During the course of this project, I also contributed to a review of methods to analyze DNA methylation data for ecological and evolutionary studies (available here). |
Past projects

While an undergraduate, I worked with Heather Watts on a series of projects examining reproductive timing in house finches. This work has recently been published in two stages. First, we report patterns of reproductive timing in wild house finches that show egg laying has advanced over the past century, occurring earlier in warmer springs (Watts, et al. 2018 IBIS). Next, we experimentally manipulated the environment of captive house finches, showing that elevated temperatures adjust the breeding-to-molt transition, but not the onset of reproduction (Watts, et al. 2018 JEB).

